




MSICare is a computational tool for MSI phenotype detection with next-generation sequencing (NGS) data. The algorithm is based on the comparison of the reads distributions between normal and tumor samples.
Mononucleotide repeats (MNR) with a length ≥ 12 base pairs (bp) were considered for analysis only if they were covered by at least 20 mapping reads in both normal and tumor samples. The total number of reads covering each candidate MNR was then normalized (arbitrary value of 100) in tumor and matched healthy tissue. For each MNR, the normalized number of reads in healthy tissue was subtracted from the normalized number of reads in tumor tissue [∆Ratio = %Tumor-%Normal] to generate an MSI index (MSI signal, MSIg) corresponding to the sum of ∆Ratio values for all candidate MNR. The ∆Ratio value was then adjusted by estimating the tumor purity (TP) for each tumor sample, with the estimated TP corresponding to the median value of the MSI signal for all MNR with a length ≥ 14 bp covered by at least 30 reads in tumor and 20 reads in normal tissue. The adjusted value for ∆Ratio was then used to classify a given MNR as wild type (∆Ratio-adjusted = ∆Ratio x Estimated TP < 50%) or mutated (∆Ratioadjusted = ∆Ratio x Estimated TP ≥ 50%) given that observed microsatellite mutations can be either heterozygous or homozygous in primary tumor samples. Finally, the MSICare score for tumor samples corresponds to the percentage of microsatellites that were mutated amongst the total number of microsatellites analyzed using this approach.
If you used this tool for your work, please cite PMID 33992635 link
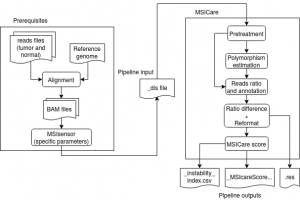
The _MSIcareScore_12_50percent.csv file reports the percentage of unstable loci as a cumulative score in the tumor and thus the MSI status.
The .res file contains detailed information on each microsatellites analysed.
Software request
Requirements
Generating a _dis file
To obtain the input of the main script you need to run MSIsensor
You first need to search the microsatellites in your reference genome
msisensor scan -d reference.fa -o microsatellites.list
Then you can obtain the coverage of the mononucleotide microsatellites of minimum length of 5 in the tumor and normal paired samples from sorted and indexed bam files.
msisensor msi -d microsatellites.list -n normal.bam -t tumor.bam -o output.prefix -p 5 -x 1 -f 10
In the folder given with -o you will find a file terminating by _dis that is the input of MSICare.sh
Software dependencies and installation
To run the main script MSICare.sh you need python2 and R (tested with version 3.5.2)
python libraries : numpy and pandas
R package : stringr
pip install numpy==1.16.3 pandas==0.24.2
R --vanilla -e "install.packages('stringr', repo='http://cran.univ-paris1.fr/')"
Usage
The script take 3 mandatory positional arguments and two optional one
MSICare.sh file_dis output_Directory path_to_scripts [ms_select] [diag]
file_dis : Path to the _dis output of MSIsensor.
MSIsensor has to be run with specific parameters
-p 5, -x 1 and -f 10 (for more details refer to the tool documentation)
outputDirectory : Path to the wanted output directory. Will be created if do not exist
path_to_scripts : Path to the folder containing the needed scripts
ms_select : Path to a file containing a list of microsatellites genomic positions
(first two columns of MSIsensor scan output separated by a tabulation) to consider
in computing the MSICare score.
By default all microsatellites passing filters are used.
diag : By default the pipeline will launch the diagnostic part. (Default T)
If you don't need it set this parameter to F
Example
- Scan microsatellites from reference genome:
msisensor scan -d reference.fa -o microsatellites.list
- Extract MSI read count distribution:
msisensor msi -d microsatellites.list -n normal.bam -t tumor.bam -e bed.file -o output.prefix -p 5 -x 1 -f 1
- MSICare scoring:
MSICare.sh file_dis outputDirectory path_to_scripts [ms_select]